18511767098


人參屬藥材因皂苷類成分的廣泛藥理活性而受到關注與研究,國內外學者研究主要集中于皂苷類成分分離與鑒定、藥理作用和質量評價等方面,亦有用液相色譜-質譜(liquid chromatography-mass spectrometry,LC-MS)聯用法對竹節參皂苷類成分進行定性分析的研究報道,共定性或鑒定出13 個皂苷類成分。竹節參中皂苷類成分常見的分析方法主要有高效液相色譜(highperformance liquid chromatography,HPLC)法、核磁共振(nuclear magnetic resonance,NMR)及LCMS等。HPLC法操作簡便、靈敏度較高,主要用于皂苷類成分的含量測定;NMR作為一種對已知或未知化合物具有較強結構解析能力的檢測手段,多用于對未知化合物的結構分析鑒定,但NMR大多需對樣品進行較復雜且耗時的提取、分離等前處理步驟;LC-MS樣品前處理簡單,而且集LC高分離能力、MS高靈敏度和高選擇性于一體,對已知或未知化合物均可較準確地分析與鑒定,已廣泛應用于中藥的定性與定量分析。
本研究采用超快速液相色譜-三重四極桿飛行時間質譜技術對竹節參中皂苷類成分進行分析,根據高分辨質譜提供的準分子離子及其產生的碎片離子精確分子質量、色譜保留時間,結合標準品比對與參考相關文獻數據,初步鑒定出原人參二醇型、原人參三醇型、齊墩果烷型及奧寇梯木醇型共53 種皂苷成分,并推測其可能的裂解途徑及規律,旨在探究竹節參的藥效物質基礎和建立其藥材品質的綜合評價體系提供基礎資料。
三七皂苷R1、人參皂苷Re、人參皂苷Rg1、擬人參皂苷F11、人參皂苷Rf、人參皂苷Rb1、人參皂苷Rg2(20S)、人參皂苷Rh1(20S)、人參皂苷Rc、人參皂苷Rh1(20R) 、人參皂苷Ro。
竹節參樣品:于2017年10月采自湖北省恩施市栽培基地(北緯30°3’49",東經109°51’41"),經南京中醫藥大學劉訓紅教授鑒定為五加科植物竹節參(Panax japonicus C.A.Mey.)的根莖。
甲醇、乙腈、甲酸(均為色譜純) 德國默克公司;甲醇(批號:143135)(色譜純) 江蘇漢邦科技有限公司;實驗用水為Milli-Q超純水。
SIL-20A XR UFLC儀 日本Shimadzu公司;Triple TOFTM5600 System-MS/MS高分辨四極桿飛行時間質譜儀(配有Peakview 1.2數據處理系統,電噴霧離子源) 美國AB Sciex公司;ME36S型電子分析天平(1/100萬)、BSA2245型電子分析天平(1/10萬) 德國賽多利斯公司;Q-500B高速多功能粉碎機 上海冰都電器有限公司;KQ-500B超聲波清洗機(500 W、40 kHz) 昆山超聲儀器有限公司;H1650-W高速離心機湖南湘儀實驗室儀器開發有限公司;Milli-Q超純水制備儀 美國Millipore公司。
色譜柱SynergiTM Hydro-RP 100 ?柱(2.0 mm×100 mm,2.5 μm);流動相0.1%甲酸-水(A)-0.1%甲酸-乙腈(B);梯度洗脫程序:0~10 min,10%~35% B;10~14 min,35%~42% B;14~24 min,42%~95% B;柱溫40 ℃;流速0.4 mL/min;進樣量2.0 μL。
電噴霧離子源負離子模式;質量掃描范圍m/z 50~1 500;離子源溫度550 ℃;氣簾氣流速40 L/min;霧化氣流速55 L/min;輔助氣流速55 L/min;噴霧電壓-4 500 V;去簇電壓-100 V。
分別取各對照品適量,精密稱定,置于5 mL容量瓶中,加70%甲醇溶液溶解制成對照品溶液。分別精密吸取對照品適量,置于10 mL容量瓶中,加70%甲醇溶液溶解制成5 μg/mL混合對照品溶液。
精密稱定樣品粉末0.5 g(過60 目篩),置于25 mL具塞錐形瓶中,加入70%甲醇溶液10 mL,密閉,稱質量,超聲處理(功率500 W,頻率40 kHz)60 min,放置冷卻,再次稱質量,用70%甲醇溶液補足質量損失,搖勻,過濾,以12 000 r/min離心10 min,取上清液,過0.22 μm微孔濾膜濾過,即得供試品溶液。
實驗數據采用Peakview 1.2軟件進行處理,裂解途徑推導圖采用ChemDraw Ultra 7.0軟件進行處理。
圖1 竹節參70%甲醇溶液提取物在負離子模式下的基峰圖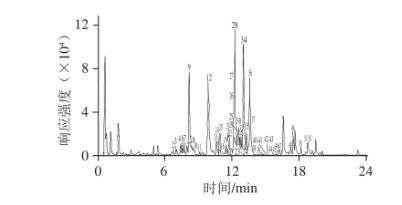
按照1.3節實驗條件對竹節參中皂苷類成分進行分離與分析,如圖1所示。通過供試品的色譜保留時間,高分辨質譜提供的準分子離子峰及碎片離子峰精確分子質量信息,與對照品比對,確定了23 個皂苷成分;根據高分辨質譜提供的精確分子質量,確定可能分子結構式,再依據碎片離子信息,參考相關文獻數據,并結合HDMB數據庫檢索,共推測出30 個皂苷成分。
圖2 竹節參中皂苷結構類型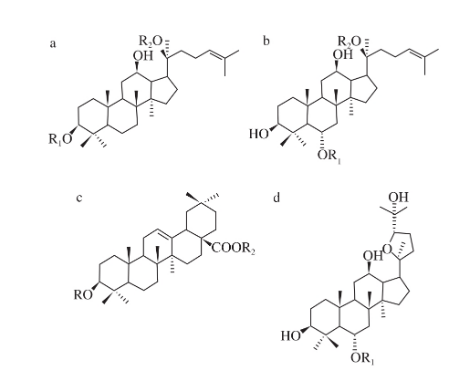
竹節參中皂苷類型主要有原人參二醇型、原人參三醇型、齊墩果烷型、奧寇梯木醇型,如圖2所示。多數皂苷含有至少一個糖鏈,一般在不同取代位置上與葡萄糖(Glc)、鼠李糖(Rha)、阿拉伯糖(Ara)、木糖(Xyl)等單糖或寡糖相連。皂苷類成分苷元部分較為穩定,一般較大壓力下也僅失去糖碎片(162、146、132 u),多數皂苷中呈現相似的單糖或寡糖碎片裂解規律。例如,人參皂苷Rb1(化合物21)的裂解碎片中除表1碎片m/z 945、783、765、621、459,還可觀察到碎片m/z 323、221、179、161、119、101,其中,m/z 323、221可以推斷為Glc-Glc裂解產生的碎片,m/z 179、161、119、101可以推斷為Glc裂解產生的碎片。
以人參皂苷Rb1、人參皂苷Rs3、人參皂苷Rg3(20R)為例說明裂解途徑。化合物21的保留時間為11.76 min,準分子離子峰為m/z 1 107.599 5[M-H]-,分別失去1、2、3、4分子Glc得到碎片離子m/z 945、783、621、459,失去2分子Glc和1分子H2O得到碎片離子m/z 765。
化合物47的保留時間為17.22 min,準分子離子峰為m/z 871.509 1[M+HCOO]-,失去C-3位上糖基連有的1分子乙酰基(Ac)得到碎片離子m/z 783,繼而失去1分子H2O得到碎片離子m/z 765,失去C-3位上糖基連有的1分子Ac、1分子Glc得到碎片離子m/z 621,再失去1分子Glc得到碎片離子m/z 459,結合文獻數據推測為人參皂苷Rs3。
以丙二酰基人參皂苷Rg1、三七皂苷R2(20S)、人參皂苷Rf為例說明裂解途徑。化合物10的保留時間8.71 min,準分子離子峰為m/z 885.488 4[M-H]-,失去C-6位上的1分子CO2得到碎片離子m/z 841,失去C-6位上糖基連有的1分子丙二酰基(Ma)得到碎片離子m/z 799,再失去1分子H2O得到碎片離子m/z 781,失去C-6位上的1分子Ma、1分子Glc得到碎片離子m/z 637,再失去1分子H2O得到碎片離子m/z 619,失去C-6位上的1分子Ma、C-6和C-20位上各1分子Glc得到苷元碎片離子m/z 475。通過與文獻數據比對推測為丙二酰基人參皂苷Rg1。
化合物15的保留時間為10.91 min,其準分子離子峰m/z 845.492 2[M+HCOO]-,分別失去C-6位上的1、2分子Glc得到碎片離子m/z 637、475。通過與對照品比對,結合文獻數據確定為人參皂苷Rf。
共鑒定了10 個齊墩果烷型皂苷,分別為表1中的化合物12、27、29、31、33、34、36、37、49和51。齊墩果烷型皂苷在一級質譜中產生較強的[M+HCOO]-、[M-H]-離子,二級質譜中C-3(R)、C-28酯基(R2)位上的糖基取代基不同程度地裂解。
以竹節參皂苷IV、竹節參皂苷IVa、齊墩果酸28-O-β-D-吡喃葡萄糖為例說明其裂解途徑。化合物34的保留時間為13.06 min,準分子離子峰為925.483 5[M-H]-,失去C-28酯基相連的1分子Glc、C-3位上與GlcUA相連的1分子Ara和1分子H2O,得到碎片離子m/z 613,再失去GlcUA上的1分子CO2得到碎片離子m/z 569。通過與對照品比對,并結合文獻數據比對后確定為竹節參皂苷IV。
共鑒定了4 個奧寇梯木醇型皂苷,分別為表1中的化合物1、2、13和14。奧寇梯木醇型皂苷在一級質譜中多產生較強的[M+HCOO]-離子,二級質譜中C-6(R1)位上的糖基取代基不同程度地裂解。
化合物2的保留時間為6.91 min,其準分子離子峰子m/z 861.486 5[M+HCOO]-,分別失去1、2分子Glc得到碎片離子m/z 653、491,珠子參苷R1的裂解途徑如圖3A所示。與文獻數據比對,推測化合物2為珠子參苷R1。
化合物1、13、14的保留時間分別為5.28、10.61、10.87 min,準分子離子峰分別為子m/z 1 023.541 4[M+HCOO]-、831.477 7[M+HCOO]-、845.493 1[M+HCOO]-,分別失去2分子Glc、1分子Xyl、1分子Rha得到碎片離子m/z 653,通過與文獻數據比對推測化合物1、13分別為葉三七皂苷B、珠子參苷R2/假人參皂苷RT2,與對照品對照并與文獻數據比對確定化合物14為擬人參皂苷F11。
通過對上述4 個奧寇梯木醇型皂苷裂解規律的推測與比對,可知奧寇梯木醇型皂苷母核較穩定,一般斷裂為糖碎片,易產生m/z 653[C36H61O10]-的碎片離子,最終可能會產生m/z 491 [C30H51O5]-的特征碎片離子,奧寇梯木醇型皂苷的裂解途徑如圖3B所示。
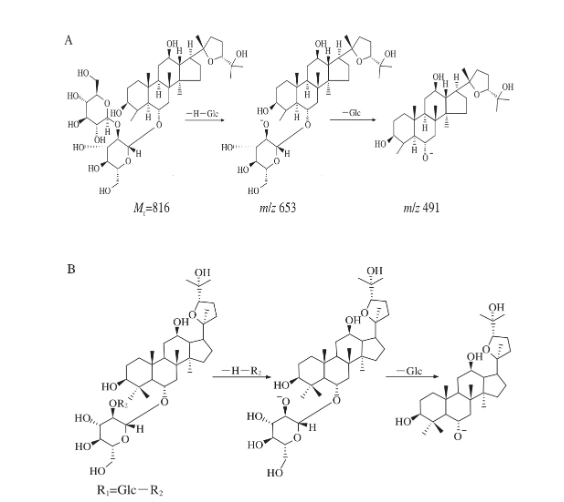
圖3 珠子參苷R1(A)和奧寇梯木醇型皂苷(B)的裂解途徑
UFLC-Triple TOF MS/MS技術可提供準分子離子及碎片離子的元素組成等結構信息,提高結構解析的準確性及分析速率,易于發現傳統分析中被遺漏或者忽略的化合物,在中藥復雜組分的鑒定研究中具有一定優勢及應用前景。但該多級質譜分析技術在結構鑒定及裂解方面仍然存在一定局限性。例如,取代基取代不同位點的同分異構體難以確認,取代基上基團斷裂位置及順序尚不能確定,裂解途徑的合理解釋等相關文獻較少。因此,采用UFLC-Triple TOF MS/MS技術對化合物結構鑒定及裂解推導仍需進一步研究,通過歸納相關化合物裂解方式及規律,為中藥中相似成分的結構分析提供理論依據。
本研究采用UFLC-Triple TOF MS/MS技術對竹節參中皂苷類成分進行分析,根據相關質譜數據、對照品及文獻,初步鑒定出原人參二醇型、原人參三醇型、齊墩果烷型及奧寇梯木醇型共53 種皂苷成分并推測其可能裂解途徑及規律,以期進一步探究竹節參藥效物質基礎及建立其品質評價體系。
技術支持:化工儀器網 管理登陸 sitemap.xml
 歡迎來到
歡迎來到